
단백질의 구조, protein structure
아미노산 사슬에 있는 원자와 분자의 3차원 배열이다. 단백질은 아미노산의 염기서열에서 형성된 생체고분자(폴리펩타이드)이다. 단일 아미노산 단량체는 중합체의 반복 단위를 나타내는 잔기라고도 할 수 있다. 단백질은 축합 반응을 겪는 아미노산에 의해 형성되는데, 이 과정에 아미노산은 펩타이드 결합으로 서로 결합하기 위해 탈수 반응을 통해 물 분자 하나씩을 잃게 된다. 관례상, 아미노산 30개 이하의 사슬은 단백질이 아닌 펩타이드로 식별되는 경우가 많다. 단백질은 생물학적 기능을 수행할 수 있도록 수소 결합, 이온 결합, 반데르발스 힘, 소수성 결합과 같은 다수의 비공유적 상호작용에 의해 구동되는 하나 이상의 특정한 공간적 형태로 접힌다. 단백질의 기능을 분자 수준에서 이해하려면, 단백질의 3차원 구조를 결정하는 것이 중요하다. 엑스선결정학, 핵자기 공명, 이중 편광 간섭법 등의 기법을 채용해 단백질의 구조를 결정한다.
단백질의 구조는 수만~수천 개의 아미노산의 크기를 갖는다. 물리적 크기에 따라 단백질은 1~100nm 사이의 나노입자로 분류된다. 단백질 소단위체로부터 매우 큰 중합체가 형성될 수 있다. 예를 들어 수천 개의 액틴이 결합해 미세섬유로 조립된다.
단백질은 일반적으로 생물학적 기능 수행에서 가역적 구조적 변화를 겪는다. 동일한 단백질의 각기 다른 구조는 다른형태 이성질체로 지칭되며, 이들 사이의 전이를 형태 변화(Comformational Change)라고 한다.
단백질의 4가지 구조
1차 구조
단백질의 1차 구조는 폴리펩타이드 사슬에서 아미노산 서열을 지칭한다. 1차 구조는 단백질 생합성 과정에서 만들어진 펩타이드 결합에 의해 유지된다. 폴리펩타이드 사슬의 두 말단은 각 말단에서 잔기의 성질에 기초하여 C 말단 및 N 말단으로 지칭된다. 잔기의 수는 항상 N 말단(NH2-그룹)에서 시작하는데, 이는 아미노기가 펩타이드 결합에 관여하지 않는 끝이다. 단백질의 1차 구조는 단백질에 해당하는 유전자에 의해 결정된다. DNA의 특정 뉴클레오타이드 서열은 전령 RNA로 전사되며, 이는 리보솜에 의해 번역이라는 과정에서 읽힌다. 인슐린의 아미노산 서열은 프레더릭 생어에 의해 발견되어 단백질에 아미노산 서열이 존재함을 확인했다. 단백질의 서열은 그 단백질에 고유하며, 단백질의 구조 및 기능을 정의한다. 단백질의 순서는 에드만 분해 또는 질량 분석법과 같은 방법으로 확인할 수 있다. 또 유전 부호를 사용하여 유전자 서열에서 직접 읽을 수도 있다. 펩타이드 결합이 형성될 때 물 분자(탈수 축합 반응)가 소실되어 단백질이 아미노기로 구성되므로, 단백질을 논의 할 때 아미노기라는 단어를 사용하는 것이 좋다. 인산화 및 당화와 같은 번역 후 변형은 일반적으로 1차 구조의 일부로 간주되며 유전자에서 읽을 수 없다. 예를 들어 인슐린은 2개의 사슬에 51개의 아미노산으로 구성되어 있다. 하나의 사슬에는 31개의 아미노산이 있고 다른 사슬에는 20개의 아미노산이 있다.
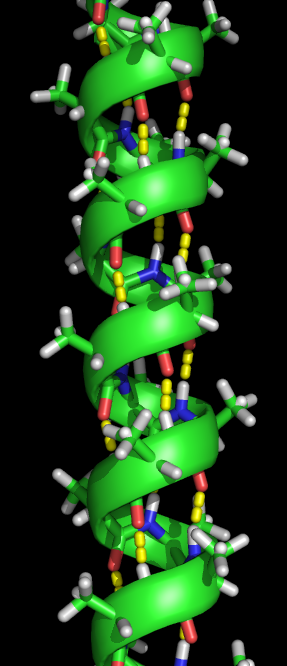
2차 구조
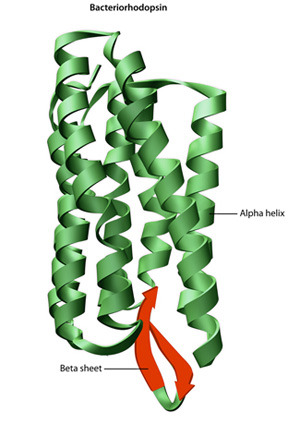
단백질의 2차 구조는 실제 폴리펩타이드 골격 사슬 상의 매우 규칙적인 국소 구조를 뜻한다. 단백질의 2차 구조인 알파 나선 구조, 베타 가닥, 베타 병풍 구조는 1951년 라이너스 폴링이 발견하였다. 이 단백질의 2차 구조는 주사슬 펩타이드 사이의 수소 결합 패턴에 의해 정의된다. 그들은 라미찬드란 조사구의 다면각 ψ 및 φ의 특정 값으로 제한되는 규칙적인 형상을 가지고 있다. 알파 나선 구조 및 베타 병풍 구조는 모두 펩타이드 골격에서 모든 수소 결합 공여체 및 수용체를 포화시키는 방법을 나타낸다. 이들은 고정된 3차원 구조가 결여된 펼쳐진 폴리펩타이드 쇄인 무작위 코일과 혼동되어서는 안된다.
3차 구조
단백질의 3차 구조는 단량체 및 다량체 단백질 분자의 3차원 구조를 뜻한다. 알파 나선 구조 및 베타 병풍 구조는 컴팩트한 구형 구조로 접혀 있다. 접힘은 비특이적 소수성 상호작용, 물로부터의 소수성 잔류물의 매장 등에 의해 추진되지만, 단백질 도메인의 부분이 염다리, 수소 결합, 잔기 및 다이설파이드 결합의 촘촘한 패킹과 같은 특정 3차 상호작용에 의해 제자리에 고정되어야만 구조가 안정된다. 세포액이 일반적으로 환원 환경이기 때문에, 다이설파이드 결합은 세포액 단백질에서는 극히 드물다.
4차 구조
단백질의 4차 구조는 단일 기능 단위(다량체)로서 작용하는 둘 이상의 개별 폴리펩타이드 사슬(서브 유닛)의 응집으로 이루어진 3차원 구조이다. 생성된 다량체는 단백질의 3차 구조에서와 동일한 비공유 상호작용 및 다이설파이드 결합에 의해 안정화된다. 둘 이상의 폴리펩타이드(다수의 서브 유닛)의 복합체를 다량체(Oligomer)라 한다. 구체적으로는 2개의 서브 유닛을 포함하면 이량체(다이머), 3개의 서브 유닛을 포함하는 경우 삼량체, 4개의 서브 유닛을 포함하는 경우 사량체(테트라머), 5개의 서브 유닛을 포함하는 경우 오량체(펜타머)로 불린다. 동일한 서브 유닛으로 구성된 중합체는 'homo-'라는 접두사를 사용하며, 서로 다른 서브 유닛으로 구성된 중합체는 'hetero-'라는 접두사를 사용한다. 예를 들어 헤모글로빈의 알파 서브 유닛 2개와 베타 체인 2개와 같은 헤테로테트라머(Heterotetramer)가 있다.
단백질 구조의 도메인, 모티프, 접힘

단백질은 종종 여러 구조 단위로 구성된 것으로 설명된다. 이러한 단위에는 단백질 도메인, 모티프, 접힘이 포함된다. 진핵생물 시스템에서 약 100,000개의 상이한 단백질이 발현된다는 사실에도 불구하고, 상이한 도메인, 구조적 모티프 및 접힘이 훨씬 적다.
구조적 도메인
구조적 도메인(Structural Domain)은 자체인 안정화와 종종 단백질 사슬의 나머지 부분과 독립적으로 접히는 단백질의 전체 구조의 요소이다.
많은 도메인 중 하나의 유전자 또는 하나의 유전자 패밀리의 단백질 산물에 고유하지 않지만 대신 다양한 단백질에 나타난다. 도메인은 그들이 속하는 단백질의 생물학적 기능을 두드러지게 나타 내기 때문에 이름이 붙여지고 분류된다. 예를 들어, 칼슘 결합 칼모듈린 도메인(Calcuim-binding doamin of calmodulin)이 그 예이다. 이들이 독립적으로 안정하기 때문에, 도메인은 하나의 단백질과 다른 단백질 사이의 유전공학에 의해 스와핑되어 융합 단백질을 생성 할 수 있다.
구조 및 서열 모티프
구조 및 서열 모티프는 다수의 상이한 단백질에서 발견된 단백질 3차원 구조 또는 아미노산 서열의 짧은 세그먼트를 지칭한다.
초 2차 구조
초 2차 구조(Supersecondary Structure)는 β-α-β 단위 또는 나선-회전-나선 모티프와 같은 2차 구조 요소의 특정 조합을 지칭한다. 이들 중 일부를 구조적 모티프라고도 한다.
단백질 접힘
단백질 접힘(Protein Folding)은 나선 다발 구조(Helix bundle), 베타 통 구조(Beta barrel), 로즈만 접힘 또는 단백질의 구조적 분류 데이터베이스에 제공된 다른 접힘과 같은 일반적인 단백질 구조를 말한다. 관련 개념은 단백질 내의 접촉의 배열을 지칭하는 단백질 위상(Protein Topology)이다.
초 도메인
초 도메인(Superdomain)은 단일 단위로 상속되고 다른 단백질에서 발생하는 둘 이상의 관련이 없는 구조 도메인으로 구성된다. 그 예로, PTEN의 단백질 티로신 인산 가수 분해 효소 도메인 및 C2 도메인 쌍, 몇몇 텐신 단백질, 옥신은 식물 및 진균의 단백질에 의해 제공된다. PTP-C2 초 도메인은 곰팡이의 발산 이전에 존재 한 것으로 보이며, 이 결과에 따라 식물과 동물의 나이는 약 15억 년이 될 것으로 보인다.
단백질 역학
단백질은 정적으로 고정된 물체가 아니라 어떠한 형태에 맞게 그 상태를 변화시킨다. 이러한 상태 사이의 전환은 일반적으로 나노 스케일 상에서 발생하며, 다른 자리 입체성 조절, 효소 촉매 분석과 같은 기능적으로 관련된 현상과 연관되어 왔다. 단백질 역학 및 구조적 변화로 단백질은 세포 내에서 나노 크기의 분자 기계로 기능할 수 있으며, 종종 단백질 복합체의 형태로 작용한다.
근육 수축을 담당하는 미오신과 같은 모터 단백질, 미세소관을 따라 핵에서 세포 내부로 소포를 이동시키는 키네신, 세포 내부로 소포를 핵 쪽으로 이동시키는 운동 섬모와 편모의 운동을 생성하는 디네인 등이 그 예다.
단백질 접힘
RNA가 번역됨에 따라, 폴리펩타이드는 무작위 코일로서 리보솜을 빠져 나와 본래 상태로 접힌다. 단백질 접힘은 폴리펩타이드에서 아미노산 사이의 상호 작용 네트워크에 의해 결정되므로, 단백질 사슬의 최종 구조는 아미노산 서열(Anfinsen's dogma)에 의해 결정된다.
단백질 안정성
단백질 안정성은 1) 비공유 정전기 상호 작용, 2) 소수성 상호 작용과 같은 몇 가지 요인에 따라 달라진다. 이 상호 작용 에너지는 20-40kJ/mol 정도이다. 단백질은 온도 변화에 매우 민감하며, 이 온도의 변화는 단백질을 펼쳐지거나 변성되게 할 수 있다. 단백질 변성은 기능 상실을 초래한다.
엑스선결정학 및 열량 측정법은 단백질의 기능 및 구조에 대한 온도 변화의 영향을 설명하는 일반적인 메커니즘이 없음을 나타낸다. 이는 단백질이 에너지 관점에서 균일한 종류의 화학 물질이 아니라는 뜻이기도 하다. 개별 단백질의 구조 및 안정성은 극성 및 비극성 잔기의 비에 의존한다. 그들은 로컬 및 비로컬 상호 작용의 형태 및 순엔탈피에 기여한다.
구조적 완전성을 담당하는 분자 간 약한 상호 작용을 고려할 때, 가상의 자유에너지 균형 및 온도 의존성에 기여하는 알려지지 않은 요소가 너무 많기 때문에 온도의 영향을 예측하기가 어렵다. 내부의 염분 결합은 열 안정성을 생성하며, 저온으로 인해 이들 결합이 불안정화되는지 여부는 알려져 있지 않다.
단백질 구조 결정
방법 및 연도에 따른 단백질 구조 결정률
단백질 정보 은행에서 이용할 수 있는 단백질 구조의 약 90%가 엑스선 결정법에 의해 결정되었다. 이 방법을 이용하면 결정화된 상태에서 단백질 내 전자의 3차원 밀도 분포를 측정 할 수 있으며, 이에 따라 모든 원자의 3차원 좌표가 특정 해상도로 결정되는 것을 유추 할 수 있다. 알려진 단백질 구조의 대략 9%가 핵 자기 공명(NMR) 기술에 의해 얻어졌다. 큰 단백질 복합체는 저온전자현미경을 이용하여 단백질 구조를 결정할 수 있다. 일반적으로 해상도는 엑스선결정학 또는 핵자기 공명의 해상도보다 낮지만 최대 해상도는 꾸준히 증가하고 있다. 이 기술은 캡시드 및 아밀로이드 섬유와 같은 매우 큰 단백질 복합체에 특히 유용하다.
일반적인 2차 구조 조성은 원형이색성을 통해 결정될 수 있다. 펩타이드, 폴리펩타이드 및 단백질의 형태를 특성화하기 위해 적외선 분광법을 이용할 수도 있다. 2차원 적외선 분광법은 다른 방법으로는 연구할 수 없는 유연한 펩타이드 및 단백질의 구조를 조사하는 유용한 방법이 되었다. 단백질 구조에 대한보다 정성적인 그림은 종종 단백질 분해에 의해 얻어지며, 이는 보다 결정 성있는 단백질 샘플을 스크리닝하는 데 유용하다. 고속 병렬 단백질 분해 (FASTpp)를 이용하여 단백질을 정제할 필요없이 구조화된 분획 및 안정성을 조사 할 수 있다. 단백질의 구조가 실험적으로 결정되면, 그 구조의 분자동역학 시뮬레이션을 사용하여 계산에 대한 추가 세부 연구가 수행된다.
Protein Structure
Proteins are the end products of the decoding process that starts with the information in cellular DNA. As workhorses of the cell, proteins compose structural and motor elements in the cell, and they serve as the catalysts for virtually every biochemical reaction that occurs in living things. This incredible array of functions derives from a startlingly simple code that specifies a hugely diverse set of structures.
In fact, each gene in cellular DNA contains the code for a unique protein structure. Not only are these proteins assembled with different amino acid sequences, but they also are held together by different bonds and folded into a variety of three-dimensional structures. The folded shape, or conformation, depends directly on the linear amino acid sequence of the protein.
What Are Proteins Made Of?
The building blocks of proteins are amino acids, which are small organic molecules that consist of an alpha (central) carbon atom linked to an amino group, a carboxyl group, a hydrogen atom, and a variable component called a side chain (see below). Within a protein, multiple amino acids are linked together by peptide bonds, thereby forming a long chain. Peptide bonds are formed by a biochemical reaction that extracts a water molecule as it joins the amino group of one amino acid to the carboxyl group of a neighboring amino acid. The linear sequence of amino acids within a protein is considered the primary structure of the protein.
Proteins are built from a set of only twenty amino acids, each of which has a unique side chain. The side chains of amino acids have different chemistries. The largest group of amino acids have nonpolar side chains. Several other amino acids have side chains with positive or negative charges, while others have polar but uncharged side chains. The chemistry of amino acid side chains is critical to protein structure because these side chains can bond with one another to hold a length of protein in a certain shape or conformation. Charged amino acid side chains can form ionic bonds, and polar amino acids are capable of forming hydrogen bonds. Hydrophobic side chains interact with each other via weak van der Waals interactions. The vast majority of bonds formed by these side chains are noncovalent. In fact, cysteines are the only amino acids capable of forming covalent bonds, which they do with their particular side chains. Because of side chain interactions, the sequence and location of amino acids in a particular protein guides where the bends and folds occur in that protein (Figure 1).
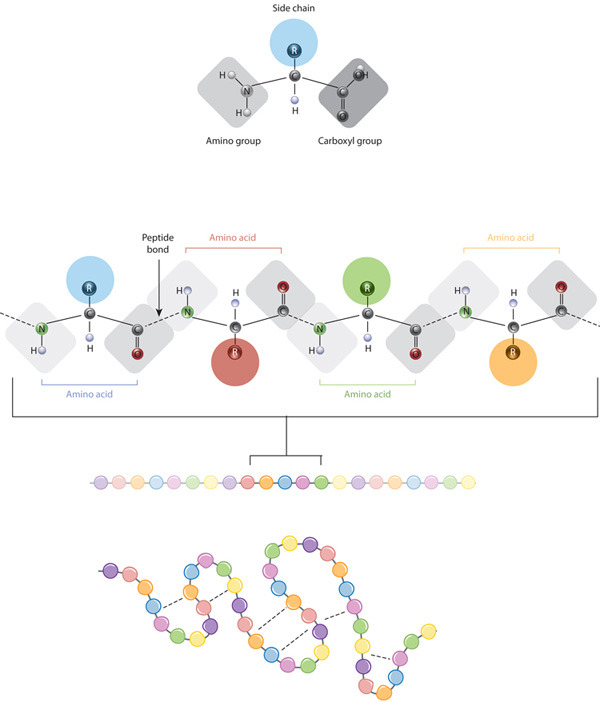


The final shape adopted by a newly synthesized protein is typically the most energetically favorable one. As proteins fold, they test a variety of conformations before reaching their final form, which is unique and compact. Folded proteins are stabilized by thousands of noncovalent bonds between amino acids. In addition, chemical forces between a protein and its immediate environment contribute to protein shape and stability. For example, the proteins that are dissolved in the cell cytoplasm have hydrophilic (water-loving) chemical groups on their surfaces, whereas their hydrophobic (water-averse) elements tend to be tucked inside. In contrast, the proteins that are inserted into the cell membranes display some hydrophobic chemical groups on their surface, specifically in those regions where the protein surface is exposed to membrane lipids. It is important to note, however, that fully folded proteins are not frozen into shape. Rather, the atoms within these proteins remain capable of making small movements.
Even though proteins are considered macromolecules, they are too small to visualize, even with a microscope. So, scientists must use indirect methods to figure out what they look like and how they are folded. The most common method used to study protein structures is X-ray crystallography. With this method, solid crystals of purified protein are placed in an X-ray beam, and the pattern of deflected X rays is used to predict the positions of the thousands of atoms within the protein crystal.
How Do Proteins Arrive at Their Final Shapes?
In theory, once their constituent amino acids are strung together, proteins attain their final shapes without any energy input. In reality, however, the cytoplasm is a crowded place, filled with many other macromolecules capable of interacting with a partially folded protein. Inappropriate associations with nearby proteins can interfere with proper folding and cause large aggregates of proteins to form in cells. Cells therefore rely on so-called chaperone proteins to prevent these inappropriate associations with unintended folding partners.
Chaperone proteins surround a protein during the folding process, sequestering the protein until folding is complete. For example, in bacteria, multiple molecules of the chaperone GroEL form a hollow chamber around proteins that are in the process of folding. Molecules of a second chaperone, GroES, then form a lid over the chamber. Eukaryotes use different families of chaperone proteins, although they function in similar ways.
Chaperone proteins are abundant in cells. These chaperones use energy from ATP to bind and release polypeptides as they go through the folding process. Chaperones also assist in the refolding of proteins in cells. Folded proteins are actually fragile structures, which can easily denature, or unfold. Although many thousands of bonds hold proteins together, most of the bonds are noncovalent and fairly weak. Even under normal circumstances, a portion of all cellular proteins are unfolded. Increasing body temperature by only a few degrees can significantly increase the rate of unfolding. When this happens, repairing existing proteins using chaperones is much more efficient than synthesizing new ones. Interestingly, cells synthesize additional chaperone proteins in response to "heat shock."
What Are Protein Families?
All proteins bind to other molecules in order to complete their tasks, and the precise function of a protein depends on the way its exposed surfaces interact with those molecules. Proteins with related shapes tend to interact with certain molecules in similar ways, and these proteins are therefore considered a protein family. The proteins within a particular family tend to perform similar functions within the cell.
Proteins from the same family also often have long stretches of similar amino acid sequences within their primary structure. These stretches have been conserved through evolution and are vital to the catalytic function of the protein. For example, cell receptor proteins contain different amino acid sequences at their binding sites, which receive chemical signals from outside the cell, but they are more similar in amino acid sequences that interact with common intracellular signaling proteins. Protein families may have many members, and they likely evolved from ancient gene duplications. These duplications led to modifications of protein functions and expanded the functional repertoire of organisms over time.
Conclusion
Proteins are built as chains of amino acids, which then fold into unique three-dimensional shapes. Bonding within protein molecules helps stabilize their structure, and the final folded forms of proteins are well-adapted for their functions.